 vectors,
vectors, 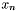 , where each has three components. As the molecular conformation changes, therefore, the result is a new set of vectors
, where each has three components. As the molecular conformation changes, therefore, the result is a new set of vectors 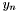 . We can capture the the differences between conformations using these two sets of vectors.
. We can capture the the differences between conformations using these two sets of vectors.If it's your day job to push proteins in silico then you will one day have to calculate the RMSD of a protein. For example, you've just simulated the protein GinormousA for a whole micro-second, but you don't even know if GinormousA is stable. Sure, you could load up a protein viewer and eyeball the stability of the protein throughout the simulation. But eye-balling is just not going to cut it with your boss -- you need to slap a number on it. This is where RMSD comes in. RMSD is a quantitative measurement of difference between two atomic structures.
A molecule's conformation is basically a set of 3-dimensional coordinates. This means that a conformation is a set of vectors, , where each has three components. As the molecular conformation changes, therefore, the result is a new set of vectors . We can capture the the differences between conformations using these two sets of vectors.
To make life easier, we first shift the center of mass of and to the origin of the coordinate system (we can always shift them back afterwards). Once "centered," the differences between the vector sets can be captured by the total squared distance between them:
![\[ E = \sum_n{|x_n - y_n|^2} \]](form_3.png)
The root mean-square deviation (RMSD) is then:
![\[ RMSD = \sqrt{\frac{E}{N}} \]](form_4.png)
But hola, you say...this mean-square measure doesn't measure similarity very well! After all, any rotation of the set of (which doesn't change the internal arrangement of ), would distort the RMSD! What we really need, then, is to find the best rotation of with respect to before taking the RMSD. To rotate the vectors , we apply a rotation matrix 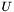 to get
to get 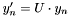 . We can then express
. We can then express 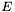 as a function of the rotation :
as a function of the rotation :
![\[ E = \sum_n{|x_n - U \cdot y_n|^2} \]](form_8.png)
Thus, to find the RMSD, we must first find the rotation 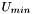 that minimizes -- the optimal superposition of the two structures.
that minimizes -- the optimal superposition of the two structures.
to get this rotation. The algorithm described in those papers have since become part of the staple diet of protein analysis. As the original proof for the Kabsch equations (using 6 different Lagrange multipliers) is rather tedious, we'll describe a much simpler proof using standard linear algebra.
First, we expand (which is now a function of ):
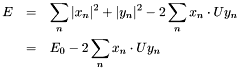
The first part, 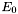 , is invariant with respect to a rotation . The variation resides in the last term,
, is invariant with respect to a rotation . The variation resides in the last term, 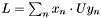 . The goal, therefore, is to find the matrix that gives the largest possible value of
. The goal, therefore, is to find the matrix that gives the largest possible value of 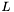 , from which we obtain:
, from which we obtain:
![\[ RMSD = \sqrt{\frac{E_{min}}{N}} = \sqrt{\frac{E_0 - 2 \cdot L_{max}}{N}} \]](form_14.png)
and into two 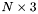 matrices
matrices 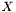 and
and 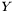 . Then we can write as a matrix trace:
. Then we can write as a matrix trace:
![\[ L = Tr( X^T U Y ) \]](form_18.png)
where 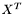 is a
is a 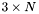 matrix and is an matrix. Juggling the matrices inside the trace, we get:
matrix and is an matrix. Juggling the matrices inside the trace, we get:
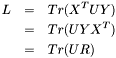
Now, we still don't know what is, but we can study the matrix 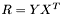 , which is the
, which is the 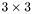 correlation matrix between and . Furthermore, by invoking the powerful Singular Value Decomposition theorem, we know that we can always write R as:
correlation matrix between and . Furthermore, by invoking the powerful Singular Value Decomposition theorem, we know that we can always write R as:
![\[ R = V S W \]](form_24.png)
and obtain the matrices 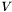 and
and 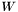 (which are orthogonal), and
(which are orthogonal), and 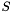 (which is positive diagonal, with elements
(which is positive diagonal, with elements 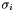 ). We can then substitute these matrices into the expression for :
). We can then substitute these matrices into the expression for :
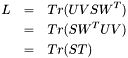
Because is diagonal, we can get a simple expression for in terms of the :
![\[ L = Tr( S T ) = \sigma_1 \cdot T_{11} + \sigma_2 \cdot T_{22} + \sigma_3 \cdot T_{33} \]](form_30.png)
. The secret sauce is in 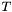 . If we look at the definition of , we can see that it is a product of orthogonal matrices, namely, the matrices ,
. If we look at the definition of , we can see that it is a product of orthogonal matrices, namely, the matrices , 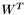 and . Thus, must also be orthogonal.
and . Thus, must also be orthogonal.
Given this, the largest possible value for any element of is 1, or 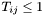 . And since is a product of terms linear in the diagonal elements of , the maximum value of occurs when the
. And since is a product of terms linear in the diagonal elements of , the maximum value of occurs when the 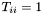 :
:
![\[ L_{max} = Tr( S T_{max} ) = \sigma_1 + \sigma_2 + \sigma_3 = Tr( S ) \]](form_35.png)
Plugging this into the equation for RMSD, we get
![\[ RMSD = \sqrt{ \frac{E_0 - 2 (\sigma_1 + \sigma_2 + \sigma_3 )}{N} } \]](form_36.png)
From this analysis, we can also determine the rotation that yields the optimal superposition of the vectors. When , then 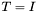 , the identity matrix. This immediately gives us an equation for the optimal rotation in terms of the left and right orthogonal vectors of the matrix R:
, the identity matrix. This immediately gives us an equation for the optimal rotation in terms of the left and right orthogonal vectors of the matrix R:
![\[ T_{max} = W^T U_{min} V = I \]](form_38.png)
![\[ U_{min} = W V^T \]](form_39.png)
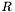 , but we still don't know how to find them! Never fear. Kabsch, who first derived the solution, showed that to solve for , we should solve the matrix
, but we still don't know how to find them! Never fear. Kabsch, who first derived the solution, showed that to solve for , we should solve the matrix 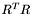 , which is a nice symmetric real matrix, and is consequently much easier to solve. Expanding on the singular value decomposition from the previous section
, which is a nice symmetric real matrix, and is consequently much easier to solve. Expanding on the singular value decomposition from the previous section
![\[ R^TR = (W^T S^T V^T)(V S W) = W^T S^2 W \]](form_42.png)
we can see that the singular value term of is 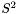 . In other words, the singular values of (
. In other words, the singular values of (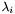 ) are related to the singular values of () by the relationship
) are related to the singular values of () by the relationship 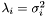 . And also, the right singular vectors are the same for both and .
. And also, the right singular vectors are the same for both and .
Furthermore, since is a symmetric real matrix, its eigenvalues are identical to its singular values . Thus, we can use any standard method (e.g. the Jacobi transformation) to find the eigenvalues of , and by substituting into the RMSD formula:
![\[ RMSD = \sqrt { \frac{ E_0 - 2 ( \sqrt{\lambda_1} + \sqrt{\lambda_2} + \sqrt{\lambda_3} )}{N} } \]](form_46.png)
from the SVD theorem. You see, is guaranteed to be orthogonal, but for our RMSD calculation, we don't just want orthogonal matrices, we want rotation matrices. And while orthogonal matrices can have determinants of +1 or -1, "proper" rotation matrices have determinants of +1 (unless they're also reflected, in which case they'll have determinants of -1, and are termed "improper" rotations).When does this happen? To answer this question, take a look at your hands. Assuming that you're a primate, you have two of them, both alike, but reflected across the plane of symmetry that runs down the center of your body. Now, try to superimpose your hands. Go ahead. Try. If you're observant, you'll notice that you simply can't superimpose your hands. Even if you cut them off of your arms, the best you can hope to do is to align them so that their outlines are coincident -- but the palm of one hand will still be touching the other. You can never perfectly superimpose your hands, because your hands are chiral.
The same thing happens in molecular systems -- molecules are frequently chiral, and when two molecules are related by chirality, it sometimes happens that their optimal superposition involves a rotation, followed by a reflection. Thus, the "improper" rotation. Unfortunately, a reflection in will result in an incorrect RMSD value using the stated algorithm, so we need to detect this situation before it becomes a problem.
To restrict the solutions of 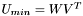 to proper rotations, we check the determinants of and :
to proper rotations, we check the determinants of and :
![\[ \omega = det( V ) \cdot det( W ) \]](form_48.png)
If 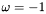 then there is a reflection in , and we must eliminate the reflection. The easiest way to do this is to reflect the smallest principal axis in , by reversing the sign of the third (smallest) eignevalue, which yields the final Kabsch formula for RMSD in terms of and
then there is a reflection in , and we must eliminate the reflection. The easiest way to do this is to reflect the smallest principal axis in , by reversing the sign of the third (smallest) eignevalue, which yields the final Kabsch formula for RMSD in terms of and 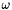 :
:
![\[ RMSD = \sqrt { \frac{ E_0 - 2 ( \sqrt{\lambda_1} + \sqrt{\lambda_2} + \omega \sqrt{\lambda_3} )}{N} } \]](form_51.png)
, rather than using an iterative approach (such as Jacobi). If only RMSD is desired (i.e. the calculation of a rotation matrix is unecessary), the get_rmsd() method is more efficient.In addition to these methods, a get_trivial_rmsd() function is provided to calculate the RMSD value between two structures without attempting to superimpose their atoms.
 1.5.1
1.5.1