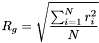
 ).
).Formally, the radius of gyration relates any N-dimensional collection of particles to an N-dimensional sphere with an equivalent moment of inertia and axis of rotation (in other words, it maps a irregular object to a sphere that will rotate the same way.) In structural biology, this is primarily useful because it is an metric for the average size, or "compacness," of a molecule.
For our purposes we'll use a slightly simpler formalism -- the radius of gyration of a molecule is the root-mean-square distance of every atom in the molecule from the molecule's geometric center:
where 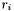 is the distance of atom
is the distance of atom  from the molecule's center, and
from the molecule's center, and  is the number of atoms in the molecule. This isn't the exact physical definition of the radius of gyration, but it works for our purposes, because the individual atomic masses rarely influence the result of the calculation.
is the number of atoms in the molecule. This isn't the exact physical definition of the radius of gyration, but it works for our purposes, because the individual atomic masses rarely influence the result of the calculation.
In order to calculate this quantity from a structure, we need to load the structure from a file (in this case, a PDB file), and to do that we need to define a few basic types:
00001 typedef BTK::ATOMS::PDBAtom atom; 00001 typedef BTK::MOLECULES::Monomer<atom> monomer; 00002 typedef BTK::MOLECULES::Polymer<monomer> polymer; 00003 typedef BTK::IO::PDBSystem<polymer> pdb_system;
These types represent a consistent set of container classes:
Once these are defined, loading the structure is trivial:
A PDBSystem is actually just a BTK::MOLECULES::System object, with the adiitional ability to parse a Protein Data Bank (PDB) structure file. In this example, we use the default behavior of the class -- it loads every chain in a PDB file, and stores them in separate Polymer chains.
Once loaded, we could use a PDBSystem to iterate over the chains in a PDB file, and then iterate over the atoms in those chains (i.e. a nested loop), but in this example, we'll make use of the ability of the PDBSystem to iterate over every atom in a loaded structure:
00001 BTK::MATH::BTKVector gc = 00002 BTK::ALGORITHMS::geometric_center(pdb.structure_begin(), 00003 pdb.structure_end());
Here we've called the geometric_center template function, which takes two AtomIterator objects that define a range of atoms, and calculates the unweighted geometric center of their positions. We then use this point to calculate the sum of squared radii for every atom in the structure:
00001 pdb_system::atom_iterator ai; 00002 double sum_rg_squared = 0.0; 00003 unsigned N = 0; 00004 00005 for (ai = pdb.structure_begin(); ai != pdb.structure_end(); ++ai) { 00006 double r = BTK::MATH::length(ai->position() - gc); // r == distance 00007 sum_rg_squared += r * r; // keep a running sum of the distance squared 00008 ++N; // count the number of atoms 00009 }
Again, we've relied upon the ability of the PDBSystem to iterate over every atom in the structure, as if they were part of a single chain. This greatly simplifies the code, as it isn't necessary to write multiple nested loops to iterate through all of the atoms in a system of polymers. Instead, we write a single, concise loop that does exactly what we need.
In this case, we have used an atom_iterator from the PDBSystem class to step through every atom in the loaded structure, and using some BTK::MATH functions, we've calulated the length (r) of the vector from the geometric center of the molecule to each atom's position. We then squared this value, and kept a running sum of the squared radii, in addition to a count of the total number of atoms in the PDBSystem (note that we could have called num_atoms() on the PDBSystem to get this value outside of the loop, if we didn't care about the extra cost of counting atoms).
Now that we have a sum of squared radii, calculation of the gyration radius of the molecule is absolutely trivial:
Here we calculate the mean squared radius (rum_rg_squared/N), and take the square root of the resulting value to obtain the gyration radius. We were able to write this (admittedly simple) application in less than 15 minutes, and in fewer than 60 lines of code, including comments. Not bad for an application that loads and parses a PDB file, does a non-trivial calculation, and produces a scientifically-useful result!
For reference, here's the complete source code for this example (which can be found in examples/gyration_radius.cpp):
00001 // -*- mode: c++; indent-tabs-mode: nil; -*- 00002 // 00003 //The Biomolecule Toolkit (BTK) is a C++ library for use in the 00004 //modeling, analysis, and design of biological macromolecules. 00005 //Copyright (C) 2006, Tim Robertson <kid50@users.sourceforge.net> 00006 // 00007 //This program is free software; you can redistribute it and/or modify 00008 //it under the terms of the GNU Lesser General Public License as published 00009 //by the Free Software Foundation; either version 2.1 of the License, or (at 00010 //your option) any later version. 00011 // 00012 //This program is distributed in the hope that it will be useful, but 00013 //WITHOUT ANY WARRANTY; without even the implied warranty of 00014 //MERCHANTABILITY or FITNESS FOR A PARTICULAR PURPOSE. See the GNU 00015 //Lesser General Public License for more details. 00016 // 00017 //You should have received a copy of the GNU Lesser General Public License 00018 //along with this program; if not, write to the Free Software Foundation, 00019 //Inc., 59 Temple Place - Suite 330, Boston, MA 02111-1307, USA. 00020 00021 #include <iostream> 00022 #include <string> 00023 #include <cmath> 00024 00025 #include <btk/core/math/vector_math.hpp> 00026 #include <btk/core/atoms/pdb_atom.hpp> 00027 #include <btk/core/molecules/monomer.hpp> 00028 #include <btk/core/molecules/polymer.hpp> 00029 #include <btk/core/io/pdb_system.hpp> 00030 #include <btk/core/algorithms/properties.hpp> 00031 00032 typedef BTK::ATOMS::PDBAtom atom; 00033 typedef BTK::MOLECULES::Monomer<atom> monomer; 00034 typedef BTK::MOLECULES::Polymer<monomer> polymer; 00035 typedef BTK::IO::PDBSystem<polymer> pdb_system; 00036 00037 int main(int argc, char * argv[]) 00038 { 00039 std::string filename; 00040 00041 if (argc == 2) { 00042 filename = argv[1]; 00043 } else { 00044 std::cerr << "usage: " << argv[0] 00045 << " <pdb filename>" 00046 << std::endl; 00047 return -1; 00048 } 00049 00050 // load the PDB file 00051 pdb_system pdb(filename); 00052 00053 // calculate the geometric center of all atoms in the PDB file 00054 BTK::MATH::BTKVector gc = 00055 BTK::ALGORITHMS::geometric_center(pdb.structure_begin(), 00056 pdb.structure_end()); 00057 00058 // iterate through every atom in the PDB file, and calculate its 00059 // distance from the geometric center of the molecule 00060 pdb_system::atom_iterator ai; 00061 double sum_rg_squared = 0.0; 00062 unsigned N = 0; 00063 00064 for (ai = pdb.structure_begin(); ai != pdb.structure_end(); ++ai) { 00065 double r = BTK::MATH::length(ai->position() - gc); // r == distance 00066 sum_rg_squared += r * r; // keep a running sum of the distance squared 00067 ++N; // count the number of atoms 00068 } 00069 00070 std::cout << "radius of gyration: " 00071 << std::sqrt(sum_rg_squared / N) << std::endl; 00072 00073 return 0; 00074 }
 1.5.1
1.5.1